Analysis pipeline
Reads to Alleles
The reads to alleles pipeline has two components:
Reads -> Genome
Genome -> Alleles
This section of the pipeline can be run locally or as part of the full pipeline on the MGT server.
When a user uploads raw paired end reads this section is run together with the rest of the pipeline.
When run locally it produces an strainID_alleles.fasta file which can be submitted to the MGT server. This approach saves upload time for large read sets and can speed up the processing time in the database. See Generating alleles file locally for more information.
Reads to Genome
Input reads are processed by kraken and samples with > 10% reads from non-target species are recorded.
Reads are then passed to the shovill pipline using SKESA for assembly.
The assembly is then filtered for quality using species specific metrics produced by assembly-stats.
If Serotyping is enabled (species dependent) SISTR is used to predict serotype from the assembly.
If the genome passed all filters it is passed to allele calling
Genome assembly to alleles
7 gene MLST ST is identified using mlst.
The genome is used to search a collection of alleles (at least one per locus) using BLASTn.
Results from this BLAST are used to extract loci from the genome if they are intact or to reconstruct the loci if they are disrupted in some way.
Extracted, reconstructed and uncallable alleles, as well as 7 gene ST, are written to a fasta file which is passed to the next stage of the pipeline.
Stages of the reads to allele pipeline are displayed in the following flow diagram.
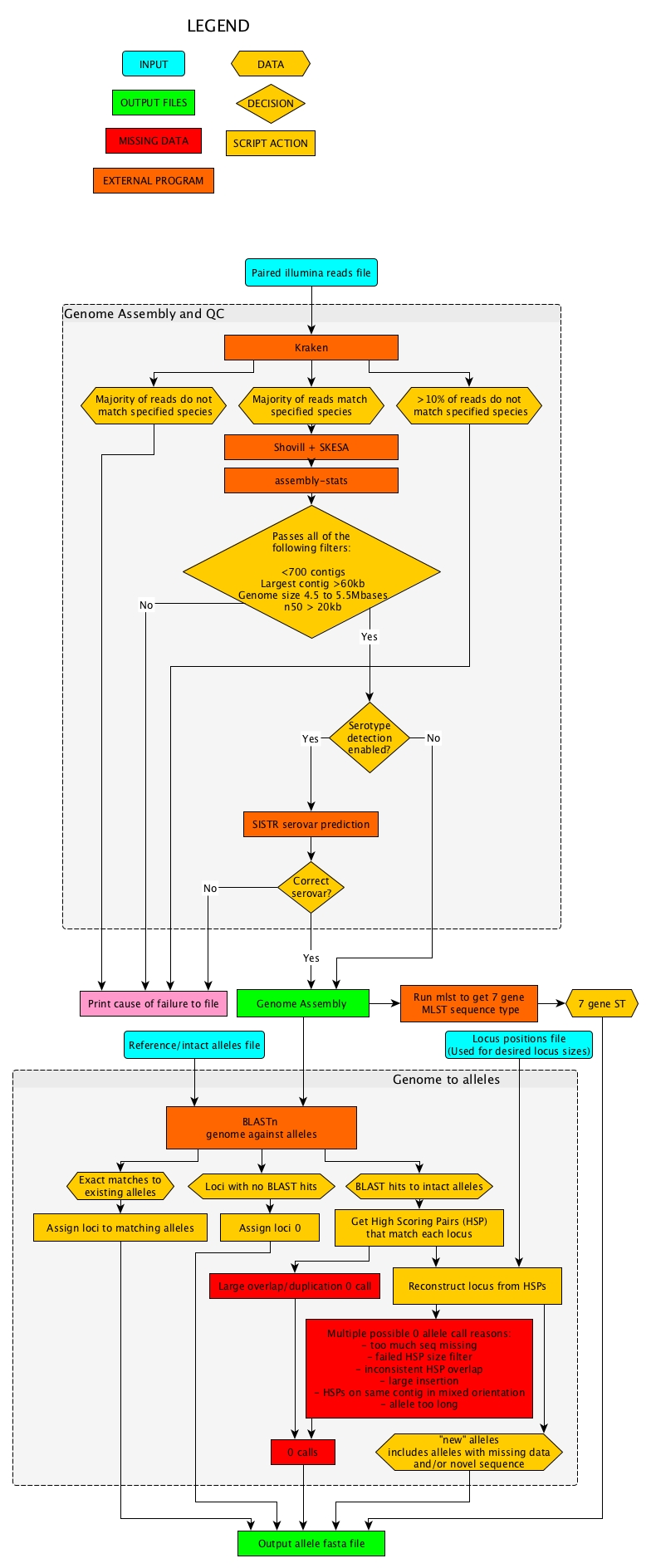
Flow diagram of Illumina reads to initial allele calls file
Alleles to MGT types
This section of the pipeline must be run in conjunction with an MGT postgres server as it reads and writes from/to the database heavily.
Stages of the alleles to MGTdb pipeline are displayed in the following flow diagram.
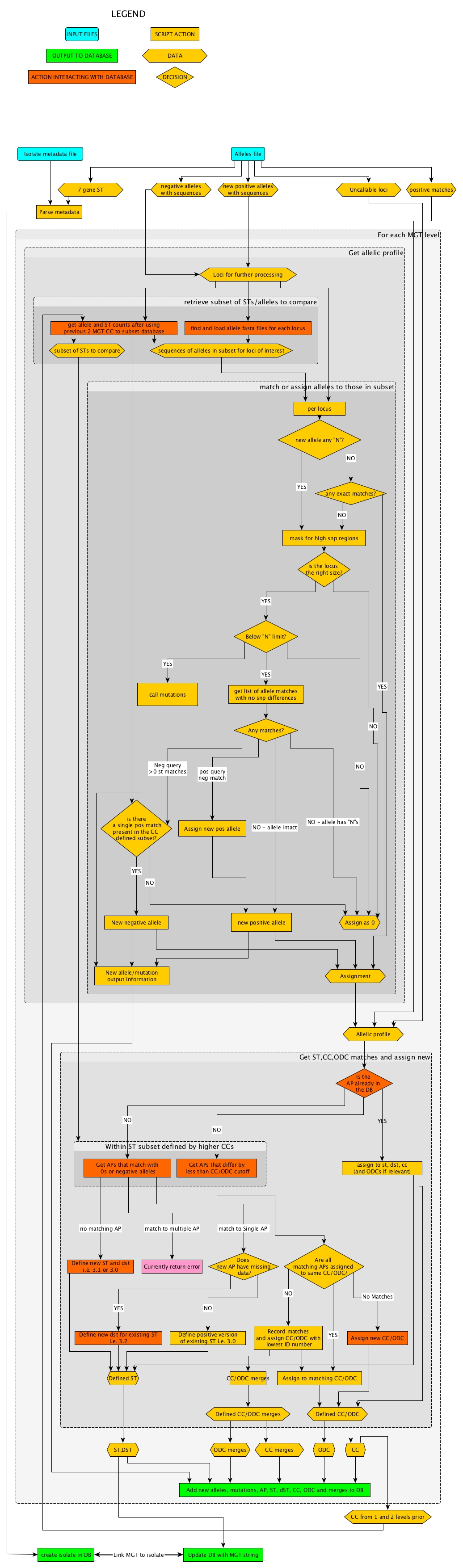
Flow diagram of initial allele calls to MGT assignments
Terminology
Negative alleles = alleles that have missing data denoted in sequence by ‘N’ may be multiple alleles with same call but different missing regions, assigned as -4_1, -4_2 for 2 different negative alleles that show no mutations from allele 4.
New positive alleles = intact alleles that do not match reference alleles (used to search in reads to alleles section).
Positive matches = alleles that are intact and match to existing reference alleles.
AP = Allele(ic) profile collection of all alleles used to define the ST for a given MGT scheme.
CC = Clonal complex (single linkage groups).
ST = Sequence types.
dST = Degenerate sequence types - sequence types that are the same as each other except for alleles with differing negative alleles or 0s i.e. ST 4 dst 1 (written 4.1) and ST 4 dst 2 (written 4.2) have APs that differ only by 0s or different negative versions of the same allele.
ODC = Outbreak Detection Clusters (single, double, 5 and 10 cutoff linkage groups derived from MGT9 allele profiles).
CCs and ODCs are assigned usign the same method with different cutoff values so they are often shown in the same processes.